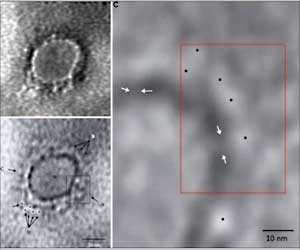
L-ORD is a rare, dominantly inherited disorder, meaning that it can occur when there is an abnormal gene from one parent. L-ORD is caused by a mutation in the gene that encodes the protein CTRP5. People with the disorder develop abnormal blood vessel growth and deposits of apolipoprotein E, which is involved in fat metabolism within the retina. Symptoms, including difficulty seeing in the dark and loss of central vision, usually appear around age 50 to 60. As L-ORD progresses, cells in the retinal pigment epithelium (RPE), a layer of tissue that nourishes the retina’s light-sensing photoreceptors, shrink and die. Loss of RPE leads to the loss of photoreceptors and in turn, to loss of vision.
The investigators were led by Kapil Bharti, Ph.D., who directs the NEI Ocular and Stem Cell Translational Research Section, and Kiyoharu (Josh) Miyagishima, Ph.D. and Ruchi Sharma, Ph.D., staff scientists in the section and leading authors of the study. They developed a laboratory model that uses induced pluripotent stem cells developed from skin (fibroblasts) to make RPE. They generated RPE from two siblings with L-ORD and, for comparison, RPE from two of the patients’ unaffected siblings who lacked the disease-causing mutation.
The patient-derived RPE shared key characteristics of the disorder in humans, including deposits of apolipoprotein E near the tissue, and abnormal secretions of vascular endothelial growth factor, a protein that stimulates blood vessel growth. The RPE cells also were dysmorphic, or deformed. By contrast, RPE from the unaffected siblings appeared normal. The researchers also found that the patient-derived RPE secreted far less of the mutant and the non-mutant CTRP5 protein compared with the models made from the unaffected siblings.
Using a computer modeling technique, they showed that mutant CTRP5 was less likely to bind with cell receptors that help fine-tune fat metabolic regulation. Less receptor binding in turn leads to chronic activation of AMP-activated protein kinase (AMPK), a key regulator of energy homeostasis and fat metabolism.
They theorized that when AMPK is chronically activated, it becomes less sensitive to imbalances in energy demand and nutrient supply. When metabolic imbalances run unchecked, they alter lipid metabolism, which explains how apolipoprotein E accumulates as deposits near the RPE layer.
Testing that theory, the researchers chemically inhibited the chronically activated AMPK in the patient-derived RPE model and found fewer deposits of apolipoprotein E, and less abnormal secretion of vascular endothelial growth factor.
Next, using the patient-derived RPE model, they tested two potential treatment strategies: A gene therapy approach to encourage expression of normal CTRP5 in the RPE model, and the use of the diabetes drug metformin, which appears to modulate AMPK activity, re-sensitizing it to changes in cellular energy status. Both strategies prevented signs of L-ORD in RPE models.
“Importantly, we now have two potential strategies to disrupt the L-ORD disease process. While gene therapy may be years away, metformin is a drug that’s long been used to treat diabetes,” said Bharti, who with NEI collaborators is planning a clinical trial to test the drug in people with L-ORD.
Although L-ORD disease is rare, it shares similarities with other retinal degenerations like age-related macular degeneration, a leading cause of vision loss. The model developed for this study may prove helpful in understanding such age-related disease changes in the RPE.
The study was funded by the NEI Intramural Research Program grants EY005121 and EY026525.
Source: Eurekalert